
O que é a Amiloidose ATTRv?
A Amiloidose Hereditária por Transtirretina (ATTRv) é uma doença sistêmica rara, autossômica dominante e decorrente de mutações no gene TTR.1
As mutações do gene TRR levam à produção de
proteína
transtirretina (TRR) instável,
que se desdobra e agrega.1
Formando fibrilas amiloides que se depositam em diversos tecidos e órgãos.1
Seu acúmulo causa disfunção progressiva, afetando principalmente o sistema nervoso periférico (ATTRv-PN) ou o coração (ATTRv-CM), ou ambos (ATTRv-Misto), embora outras manifestações sistêmicas sejam comuns.1
Focos endêmicos1-3
Portugal, Brasil, Japão e Suécia
- Foram observadas mais de 130 mutações patogênicas no gene TTR
- Val30Met é a mais comum
Além desses, casos foram relatados em outros países e mutações não-Val30Met foram frequentemente observadas.1-3
No Brasil, as mutações mais prevalentes são: Val30Met, Val122lle e Ile107Val.2
Variantes TTR

Adaptado de Marques Júnior W et al.

Sinais de alerta da ATTRv
A amiloidose ATTRv é frequentemente desconhecida ou diagnosticada tardiamente, devido à apresentação multissistêmica, heterogênea e não específica da doença. À medida que a doença progride, os sintomas e manifestações clínicas da amiloidose ATTRv frequentemente são similares aos de outras doenças mais comuns, complicando e atrasando ainda mais o diagnóstico.4,5
Os depósitos de amiloide TTR podem levar a danos progressivos em órgãos multissistêmicos5
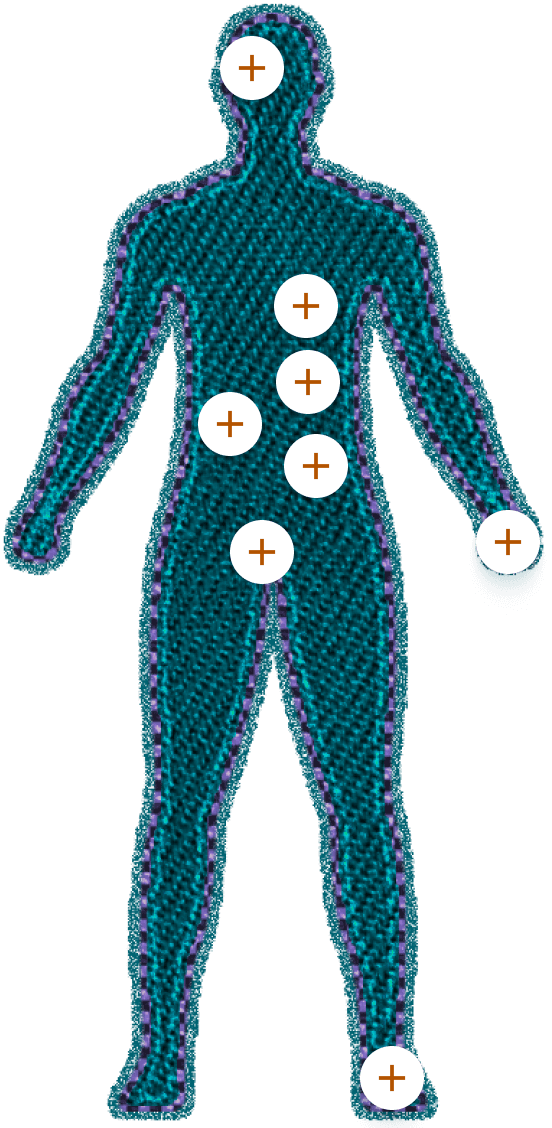
- Opacificação vítrea
- Glaucoma
- Vasos conjuntivais anormais
- Náuseas e vômitos
- Saciedade precoce
- Diarreia
- Constipação severa
- Episódios alternados de diarreia e constipação
- Perda de peso não intencional
- Hipotensão ortostática
- Infecções recorrentes do trato urinário
- Disfunção sexual
- Anormalidades na sudorese
- Bloqueios de condução
- Cardiomiopatia
- Arritmia
- Regurgitação leve
- Tipicamente axonal, fibra-dependente, simétrica e progressivamente implacável de distal para proximal.
- Opacificação vítrea
- Glaucoma
- Vasos conjuntivais anormais
- Náuseas e vômitos
- Saciedade precoce
- Diarreia
- Constipação severa
- Episódios alternados de diarreia e constipação
- Perda de peso não intencional
- Hipotensão ortostática
- Infecções recorrentes do trato urinário
- Disfunção sexual
- Anormalidades na sudorese
- Bloqueios de condução
- Cardiomiopatia
- Arritmia
- Regurgitação leve
- Tipicamente axonal, fibra-dependente, simétrica e progressivamente implacável de distal para proximal.
Adaptado de Conceição I et al.

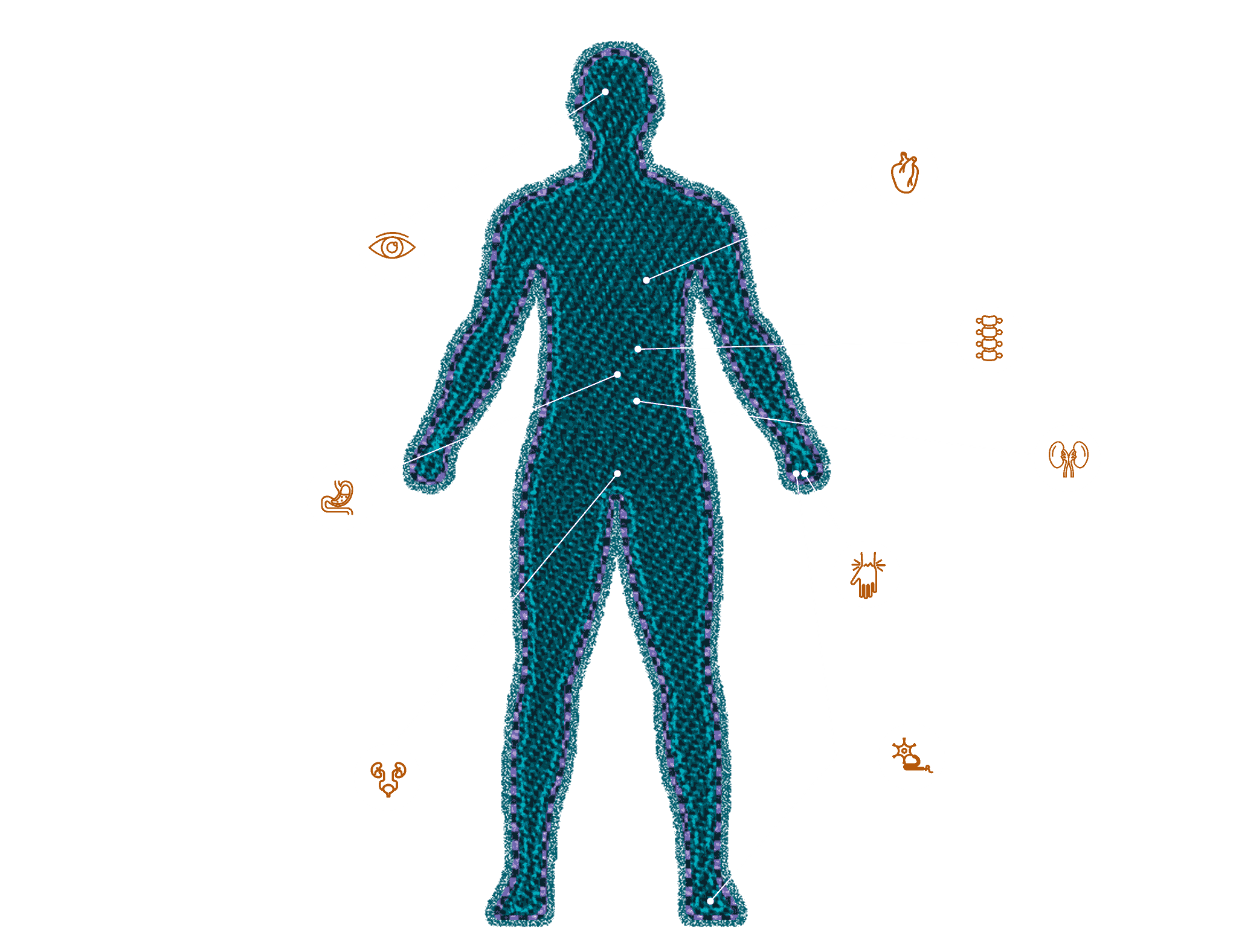
Sinais e sintomas manifestados mais comuns:4,6,7
Insuficiência cardíaca
Neuropatia periférica
Red Flags4,6,7
Comprometimento de múltiplos órgãos
Disautonomia
Síndrome do túnel do carpo bilateral
Estenose espinhal lombar
Tendão do bíceps rompido (sinal do Popeye)
Diarreia e constipação alternadas
Neuropatia periférica inexplicável
Sinais cardíacos
- Discordância entre a voltagem do ECG e a espessura da parede ventricular
- Hipotensão ortostática
- Intolerância a medicamentos padrões para insuficiência cardíaca (por exemplo, iECAs e betabloqueadores)
- Estenose aórtica paradoxal de baixo fluxo/baixo gradiente
- Persistência de níveis elevados de troponina
Embora a apresentação clínica da amiloidose ATTRv possa ser cardiomiopatia ou polineuropatia dominante, a maioria das mutações TTR dá origem a um fenótipo clínico misto, onde tanto deficiências neurológicas quanto cardíacas estão presentes.4,8,9
Polineuropatia
- Neuropatia dolorosa nas mãos e nos pés
- Fraqueza muscular
- Dificuldade para andar
Disfunção autonômica
- Diarreia crônica/
constipação/perda
de peso - Disfunção erétil
- Hipotensão/
intolerância ortostática a medicamentos para PA8
Musculoesquelético
- Ruptura do tendão do bíceps distal (sinal do Popeye)
- Estenose espinhal lombar
- Síndrome do túnel do carpo
Cardíaca
- Insuficiência cardíaca
- Fibrilação atrial
- Anormalidades de condução
O fenótipo da doença tem impacto na mortalidade dos pacientes:
- Neuropatia dominante: sobrevida mediana de 5 a 15 anos desde o diagnóstico.9
- Amiloidose ATTRv com cardiomiopatia: sobrevida mediana de 2,5 a 4 anos.9
- Ambos os fenótipos estão associados a
um
comprometimento substancial na
qualidade de vida (QV).9
Diagnóstico ATTRv com polineuropatia
Na amiloidose por transtirretina com polineuropatia ATTRv-PN, a fibrila amiloide é depositada no endoneuro. É a polineuropatia hereditária mais grave de início na idade adulta e pode envolver o coração, bem como outros órgãos.10
Seu diagnóstico é um desafio para o neurologista e demais especialidades médicas, e frequentemente atrasado, o que impacta o prognóstico funcional e vital dos pacientes. Atrasos no diagnóstico ocorrem por múltiplas razões, mas muitas vezes diagnósticos enganosos são feitos devido aos padrões de apresentação clínica esporádicos, de início tardio e altamente variados.10
Principais diagnósticos incorretos e sinais de alerta10
%
enganosas
vermelhas
- SM 4 membros
- Arreflexia difusa
- Dissociação albuminocitológica
- Desmielinização na biópsia
- ECN desmielinizante
- Dor, perda sensorial (pulsos)
- Disfunção autonômica
- Fraqueza dos membros superiores
- ECN
- Neuropatia axonal em idosos, aparentemente idiopática
- Gravidade, incapacidade rápida
- Dificuldades para caminhar
- Parestesias nas mãos
- Sem alívio após a cirurgia
- Dificuldade progressiva para caminhar em idosos
- Estenose espinhal na TC ou RM lombar
- NCS anormal
- Piora apesar da cirurgia
motor (neuropatia
motora ELA)
- Amiotrofia de membros superiores e língua
- Disartria
- Fraqueza nas mãos
- SNAP sensorial anormal (NCS)
- Nenhum sintoma de envolvimento do neurônio motor superior
- PN dependente do comprimento da fibra pequena
- Alcoolismo
- PN dependente do comprimento da fibra pequena
- Disfunção autonômica
- Rápida gravidade/duração do diabetes
- Dificuldades para caminhar
- Perda sensorial não dependente do comprimento + ataxia
- Perda de peso, nenhum anticorpo anti-onconeuronal
- Resultados negativos em PET de corpo inteiro
Adaptado de: Adams D et al.
Recomendações de consenso de especialistas para melhorar o diagnóstico de ATTRv-PN em áreas endêmicas10


(por exemplo, queixas gastrointestinais [diarreia crônica, constipação ou ambas], disfunção erétil, hipotensão postural)




Da suspeita à confirmação10
A confirmação do diagnóstico da amiloidose ATTRv-PN deve incluir teste de DNA e, em alguns casos, biópsia e tipagem amiloide.
Biópsia de deposição de amiloide
- Possíveis locais de biópsia: glândula salivar labial, tecido adiposo subcutâneo da parede abdominal, pele, rim, nervo e trato gastrointestinal (incluindo submucosa)
- Coloração vermelho-congo com birrefringência verde característica sob luz polarizada
Tipagem amiloide
- Imuno-histoquímica ou espectrometria de massa
Sequenciamento de DNA
- Análise da variante amiloidogênica TTR
- Pode dar suporte ou descartar um diagnóstico de ATTRv

O papel da genética
A amiloidose ATTRv é uma doença autossômica dominante. Portanto,
os familiares do paciente também correm o risco de ter herdado
as
mutações do gene TTR.11
O filho de um indivíduo afetado (que é heterozigoto para uma
variante TTR patogênica) tem 50% de chance de herdar a variante.11
Assim, o teste genético preditivo permite determinar se os familiares
são portadores das mutações TTR patogênicas e correm o risco de
desenvolver amiloidose ATTRv ou não.11
Os portadores assintomáticos de mutação genética, maiores de 18 anos e que demonstrem interesse, podem receber aconselhamento genético de acompanhamento anual.
Nessa sessão de aconselhamento, deve ser examinado:
O grau de compreensão e
aceitação do significado de
portar
uma mutação TTR
patogênica.
Quaisquer problemas psicológicos identificados devem ser abordados e informações sobre as opções de tratamento mais recentes disponíveis devem ser fornecidas.11
Os portadores assintomáticos de mutação genética também devem realizar exames periódicos para detecção precoce da amiloidose, especialmente devido à possibilidade de antecipação genética. O monitoramento deve começar antes da idade de início da doença observada na família.11
O médico avaliará o caso de cada paciente individualmente, podendo incluir nas avaliações periódicas:11
Entrevista médica
Exame
físico
Biópsia
e/ou
eletroneuromiografia
Exames
de sangue, urina,
oftalmológico e cardíaco

Acompanhamento holístico do paciente
A natureza progressiva da ATTRv exige monitoramento pré e pós-início da doença. Como ela pode progredir lenta ou moderadamente, é essencial que médicos de diversas especialidades estejam atentos aos primeiros sinais e sintomas da doença.3
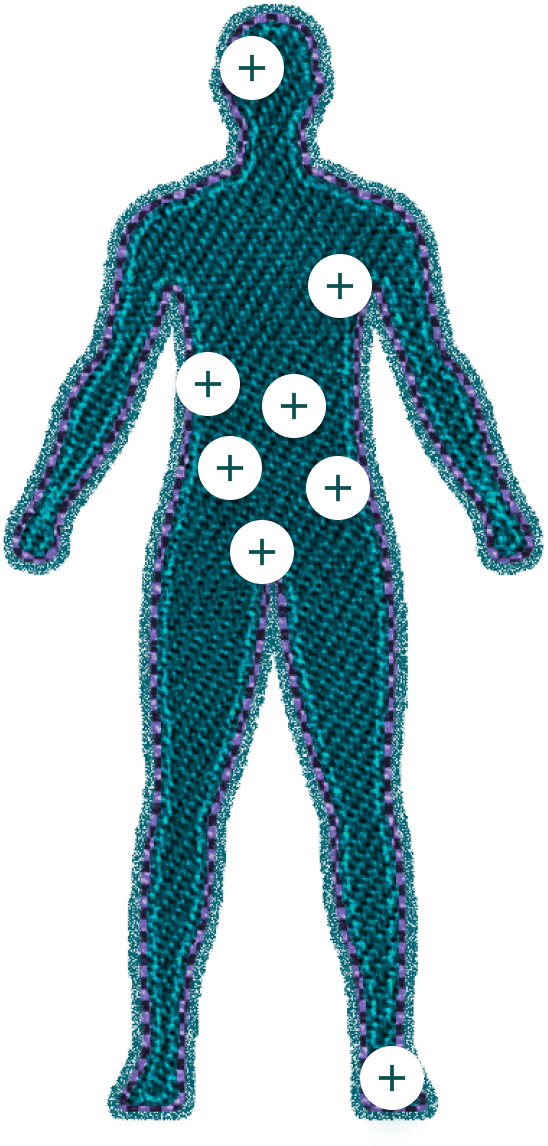
(a cada 6–12 meses)
- Olho Seco: avaliação da produção lacrimal e conforto ocular.
- Opacidades Vítreas: verifica a presença de "moscas volantes" ou outras perturbações visuais.
- Glaucoma: rastreamento para aumento da pressão intraocular.
(a cada 6 meses)
- TFGe: taxa de filtração glomerular estimada.
- Proteinúria: verifica a presença de proteína na urina.
(a cada 6 meses)
- mBMI: avaliação do Índice de Massa Corporal modificado.
(a cada 6–12 meses)
- Visão Turva: avaliação da clareza visual.
- Função Cardíaca: avalia falta de ar e palpitações.
- Função Gastrointestinal: observa náuseas/vômitos, diarreia e perda de peso não intencional.
- Avaliação de Membros Inferiores: verifica anormalidades sensoriais, fraqueza muscular, temperatura e anormalidades na sensação de dor.
- Avaliação da Síndrome do Túnel do Carpo.
(a cada 6–12 meses)
- Ecocardiograma: espessura da parede ventricular.
- ECG: monitora baixa voltagem e arritmia.
- Biomarcadores: BNP/NT-proBNP.
- Classe Funcional NYHA: avalia capacidade funcional.
(a cada 6–12 meses)
- Estudo de Condução Nervosa: CMAP/SNAP.
(a cada 6 meses)
- Hipotensão Ortostática: avalia as mudanças na pressão arterial ao ficar em pé.
- Distúrbio Urinário: avalia a função urinária.
- Disfunção Erétil: avaliação da saúde sexual.
- Sudorese Anormal: verifica a desregulação na sudorese.
(a cada 6–12 meses)
- Norfolk QoL-DN: qualidade de vida na neuropatia diabética.
- EQ-5D: um instrumento padronizado para medir resultados de saúde.
- COMPASS-31: escore composto de sintomas autonômicos.

(a cada 6–12 meses)
- Olho Seco: avaliação da produção lacrimal e conforto ocular.
- Opacidades Vítreas: verifica a presença de "moscas volantes" ou outras perturbações visuais.
- Glaucoma: rastreamento para aumento da pressão intraocular.
(a cada 6 meses)
- TFGe: taxa de filtração glomerular estimada.
- Proteinúria: verifica a presença de proteína na urina.
(a cada 6 meses)
- mBMI: avaliação do Índice de Massa Corporal modificado.
(a cada 6–12 meses)
- Visão Turva: avaliação da clareza visual.
- Função Cardíaca: avalia falta de ar e palpitações.
- Função Gastrointestinal: observa náuseas/vômitos, diarreia e perda de peso não intencional.
- Avaliação de Membros Inferiores: verifica anormalidades sensoriais, fraqueza muscular, temperatura e anormalidades na sensação de dor.
- Avaliação da Síndrome do Túnel do Carpo.
(a cada 6–12 meses)
- Ecocardiograma: espessura da parede ventricular.
- ECG: monitora baixa voltagem e arritmia.
- Biomarcadores: BNP/NT-proBNP.
- Classe Funcional NYHA: avalia capacidade funcional.
(a cada 6–12 meses)
- Estudo de Condução Nervosa: CMAP/SNAP.
(a cada 6 meses)
- Hipotensão Ortostática: avalia as mudanças na pressão arterial ao ficar em pé.
- Distúrbio Urinário: avalia a função urinária.
- Disfunção Erétil: avaliação da saúde sexual.
- Sudorese Anormal: verifica a desregulação na sudorese.
(a cada 6–12 meses)
- Norfolk QoL-DN: qualidade de vida na neuropatia diabética.
- EQ-5D: um instrumento padronizado para medir resultados de saúde.
- COMPASS-31: escore composto de sintomas autonômicos.
Adaptado de Ando Y et al.

Conheça
Até recentemente, as opções terapêuticas para ATTRv eram limitadas e consistiam
principalmente em transplante de fígado e estabilizadores de TTR. Porém, nos últimos anos, a
doença tem estado no
centro de grandes avanços terapêuticos, e o
cenário do tratamento está
mudando rapidamente, com terapias que visam aos diferentes estágios da doença,
incluindo:14-16
Silenciadores TTR
Suprimir os níveis de proteína TTR em sua fonte de produção, silenciando o gene no fígado com ASO ou siRNA.
Estabilizadores TTR
Aumentam a estabilidade do tetrâmero na circulação de TTR, evitando assim a dissociação em monômeros que podem formar fibrilas amiloides.
WAINUATM (eplontersena) é um silenciador de nova geração que usa a
tecnologia ASO
(Oligonucleotídeo Antisense) para o tratamento da amiloidose ATTRv em pacientes
adultos com
polineuropatia em estágio 1 ou 2. Funciona direcionando e ligando-se ao RNAm TTR, levando à
sua degradação e, consequentemente, reduzindo os níveis séricos de proteína TTR que podem
se
depositar nos tecidos.17-19
WAINUATM (eplontersena) foi avaliada em pacientes com ATTRv-PN no estudo principal de Fase 3 Neuro-TTRansform, que demonstrou redução significativa e sustentada na concentração de TTR em comparação com placebo.12,20
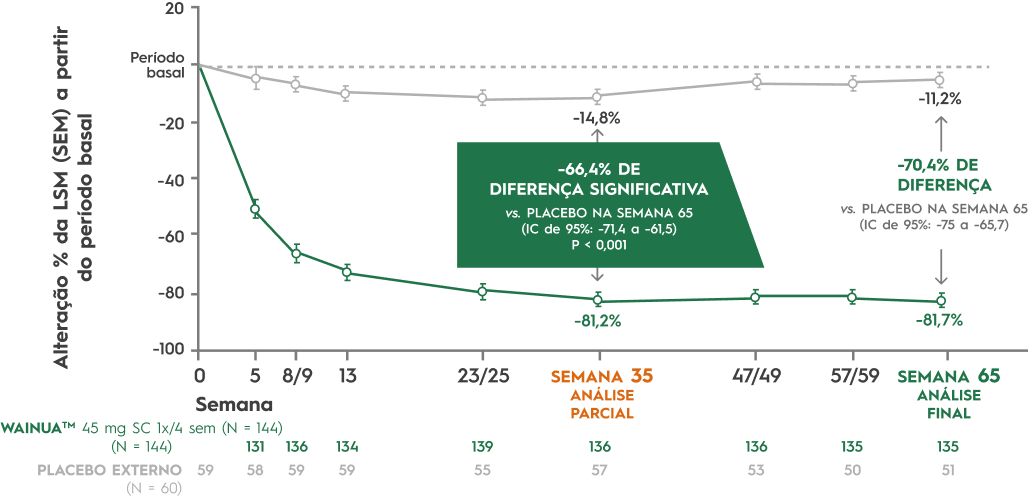
Adaptado de Coelho T el al. JAMA, 2023 Oct 17;330(15); 1448-1458.
Além disso, WAINUATM (eplontersena) interrompeu a progressão do comprometimento da neuropatia e melhorou significativamente a qualidade de vida.12,20

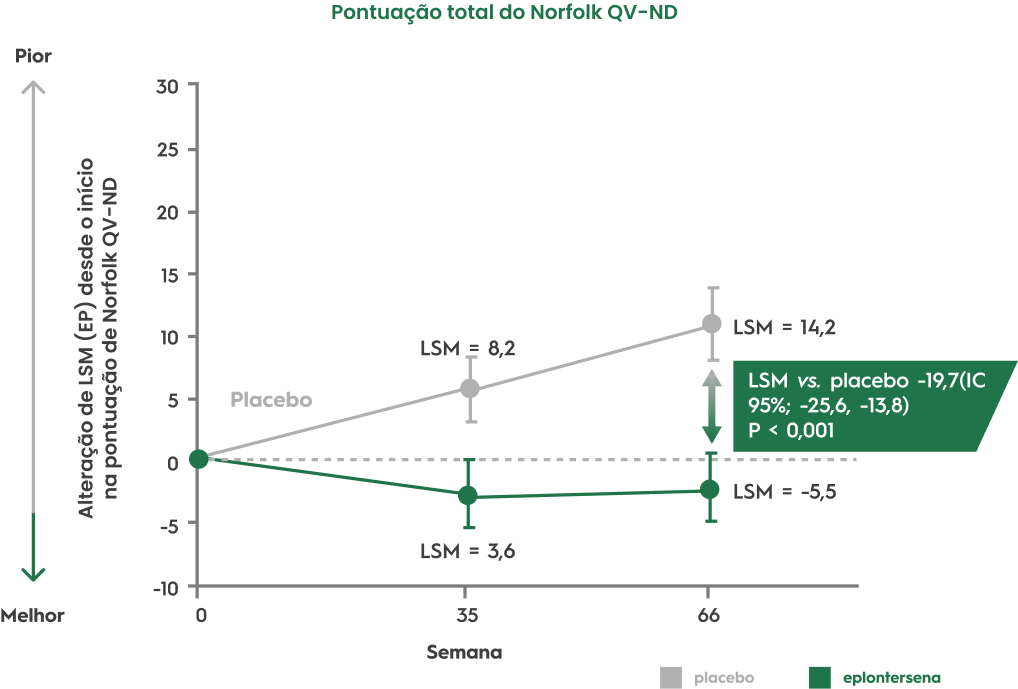
Adaptado de Conceição I et al. November 2–3, 2023; Madrid, Spain.
Simplicidade de uma dose mensal, subcutânea e autoadministrável.19
WAINUATM (eplontersena) é
autoadministrável, proporcionando flexibilidade para pacientes e cuidadores.

Formulação:
solução parenteral estéril, sem conservantes, para administração subcutânea.
Apresentação:
autoaplicador de dose única de 0,8 mL para autoadministração.
Dose:
injeção mensal de 45 mg.
Características:
dose automatizada, agulha escondida da vista, escudo de segurança da agulha após a dosagem.
Acesse a bula de WainuaTM clicando aqui
interrompe a progressão da neuropatia
e
melhora a qualidade de
vida.12,13
ELA, esclerose lateral amiotrófica; PDIC, polineuropatia desmielinizante inflamatória crônica; TC, tomografia computadorizada; STC, síndrome do túnel do carpo; RM, ressonância magnética; ECN, estudo de condução nervosa; PET, tomografia por emissão de pósitrons; PNP, polineuropatia; NP, neuropatia periférica; SM, sensório-motor; SNAP, potencial de ação nervosa sensorial (do inglês Sensory Nerve Action Potential).
Referências bibliográficas:
- Pinto MV, França MC Jr, Gonçalves MVM et al. Brazilian consensus for diagnosis, management and treatment of hereditary transthyretin amyloidosis with peripheral neuropathy: second edition. Arq Neuropsiquiatr. 2023 Mar;81(3):308-321.
- Marques Júnior W, Costa MCM, Anna Paula Covaleski AP et al. ATTRv distribution in a continental multiracial country. XIX International Symposium on Amyloidosis Abstracts. Abstract 439. Disponível em: https://www.tandfonline.com/doi/full/10.1080/13506129.2024.2347124. Acessado em 20 de maio de 2025.
- Ando Y, Waddington-Cruz M, Sekijima Y et al. Optimal practices for the management of hereditary transthyretin amyloidosis: real-world experience from Japan, Brazil, and Portugal. Orphanet J Rare Dis. 2023 Oct 12;18(1):323.
- Nativi-Nicolau JN, Karam C, Khella S et al. Screening for ATTR amyloidosis in the clinic: overlapping disorders, misdiagnosis, and multiorgan awareness. Heart Fail Rev. 2022 May;27(3):785-793.
- Conceição I, González-Duarte A, Obici L et al. "Red-flag" symptom clusters in transthyretin familial amyloid polyneuropathy. J Peripher Nerv Syst. 2016 Mar;21(1):5-9.
- Garcia-Pavia P, Rapezzi C, Adler Y et al. Diagnosis and treatment of cardiac amyloidosis: a position statement of the ESC Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2021 Apr 21;42(16):1554-1568.
- Benson MD, Dasgupta NR, Rao R. Diagnosis and Screening of Patients with Hereditary Transthyretin Amyloidosis (hATTR): Current Strategies and Guidelines. Ther Clin Risk Manag. 2020 Aug 14;16:749-758.
- Coelho T, Maia LF, Martins da Silva A et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology. 2012 Aug 21;79(8):785-92.
- Hawkins PN, Ando Y, Dispenzeri A et al. Evolving landscape in the management of transthyretin amyloidosis. Ann Med. 2015;47(8):625-38.
- Adams D, Ando Y, Beirão JM et al. Expert consensus recommendations to improve diagnosis of ATTR amyloidosis with polyneuropathy. J Neurol. 2021 Jun;268(6):2109-2122.
- Ueda M, Sekijima Y, Koike H et al. Monitoring of asymptomatic family members at risk of hereditary transthyretin amyloidosis for early intervention with disease-modifying therapies. J Neurol Sci. 2020 Jul 15;414:116813.
- Coelho T et al. Eplontersen for Hereditary Transthyretin Amyloidosis With Polyneuropathy. JAMA. 2023 Oct 17;330(15):1448-1458.
- https://www.wainua.com/, acesso em 12.03.25.
- Echaniz-Laguna A, Cauquil C, Labeyrie C, Adams D. Treating hereditary transthyretin amyloidosis: Present & future challenges. Rev Neurol (Paris). 2023 Jan-Feb;179(1-2):30-34.
- Ando Y, Adams D, Benson MD et al. Guidelines and new directions in the therapy and monitoring of ATTRv amyloidosis. Amyloid. 2022 Sep;29(3):143-155.
- Quarta CC, Fontana M, Damy T et al. Changing paradigm in the treatment of amyloidosis: From disease-modifying drugs to anti-fibril therapy. Front Cardiovasc Med. 2022 Dec 20;9:1073503.
- Nie T. Eplontersen: First Approval. Drugs. 2024 Apr;84(4):473-478. Erratum in: Drugs. 2024 Apr;84(4):489.
- Viney NJ, Guo S, Tai LJ et al. Ligand conjugated antisense oligonucleotide for the treatment of transthyretin amyloidosis: preclinical and phase 1 data. ESC Heart Fail. 2021 Feb;8(1):652-661.
- Bula de eplontersena. AstraZeneca do Brasil Ltda.
- Coelho T, Ando Y, Benson MD et al. Design and Rationale of the Global Phase 3 NEUROTTRansform Study of Antisense Oligonucleotide AKCEA-TTR-LRx (ION-682884-CS3) in Hereditary Transthyretin-Mediated Amyloid Polyneuropathy. Neurol Ther. 2021 Jun;10(1):375-389.